GTF {PrimerSeq Details}
Overview
The Gene Transfer Format (GTF) is a widely used format for storing gene annotations. You can obtain GTF files easily from the UCSC table browser and Ensembl. For example, the first few lines of UCSC’s gene annotation for hg19 looks like the following:
chr1 hg19_knownGene exon 11874 12227 0.000000 + . gene_id "uc001aaa.3"; transcript_id "uc001aaa.3";
chr1 hg19_knownGene exon 12613 12721 0.000000 + . gene_id "uc001aaa.3"; transcript_id "uc001aaa.3";
chr1 hg19_knownGene exon 13221 14409 0.000000 + . gene_id "uc001aaa.3"; transcript_id "uc001aaa.3";
chr1 hg19_knownGene exon 11874 12227 0.000000 + . gene_id "uc010nxr.1"; transcript_id "uc010nxr.1";
chr1 hg19_knownGene exon 12646 12697 0.000000 + . gene_id "uc010nxr.1"; transcript_id "uc010nxr.1";
chr1 hg19_knownGene exon 13221 14409 0.000000 + . gene_id "uc010nxr.1"; transcript_id "uc010nxr.1";
chr1 hg19_knownGene start_codon 12190 12192 0.000000 + . gene_id "uc010nxq.1"; transcript_id "uc010nxq.1";
chr1 hg19_knownGene CDS 12190 12227 0.000000 + 0 gene_id "uc010nxq.1"; transcript_id "uc010nxq.1";
chr1 hg19_knownGene exon 11874 12227 0.000000 + . gene_id "uc010nxq.1"; transcript_id "uc010nxq.1";
chr1 hg19_knownGene CDS 12595 12721 0.000000 + 1 gene_id "uc010nxq.1"; transcript_id "uc010nxq.1";
The columns are tab separated and are defined by the GTF standard specified here. PrimerSeq only uses the lines with the “exon” feature (column 3) and ignores other lines.
Be Careful
If you are particularly observant, you will notice that the above example GTF from UCSC known gene annotation has the same gene ID as transcript ID. This is because the GTF format requires a gene ID attribute, so UCSC just fills in a wrong value (the transcript ID). This example GTF and many GTFs are also not sorted by chromosome and position. PrimerSeq requires a sorted GTF as input.
I have listed ways to obtain sorted GTF files below, in suggested order.
1. Pre-sorted GTF files
If you are using a common annotation I strongly suggest you download it from the list below.
- Homo_sapiens.knownGene.hg38.sorted.withGenes.gtf
- Homo_sapiens.knownGene.hg19.sorted.withGenes.gtf
- Homo_sapiens.GRCh37.69.sorted.gtf
- Mus_musculus.knownGene.mm9.sorted.withGenes.gtf
- Mus_musculus.GRCm38.78.sorted.gtf
- Mus_musculus.GRCm38.69.sorted.gtf
- Flybase Drosophila melanogaster (r6.08)
If you feel a widely used annotation is missing, feel free to suggest that I include it by email.
2. Sorting a GTF file from the command line
Since most tasks dealing with RNA-Seq data analysis are ran from a server, I have provided a python script to sort a GTF file. An example usage scenario for a GTF file called “your_gtf_file.gtf” is shown below:
$ wget --no-check-certificate https://raw.github.com/ctokheim/PrimerSeq/master/gtf.py -O gtf.py # get command line script
$ python gtf.py -c your_gtf_file.gtf # check if GTF is sorted
your_gtf_file.gtf is not correctly sorted. please sort before use.
$ python gtf.py -i your_gtf_file.gtf -o your_gtf_file.sorted.gtf # GTF was not sorted, so sort it
3. Sorting a GTF file from PrimerSeq
PrimerSeq also includes the ability to handle such ill-specified GTF files. For any GTF file not downloaded from the PrimerSeq website, you should sort the GTF by Edit -> Sort GTF in the PrimerSeq GUI.
Warning!
Your GTF must be sorted.Do not assume so unless
you download the GTF from
this website.
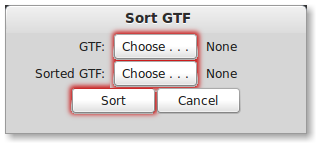
Select the GTF you wish to sort using the “GTF:” button. Next select the output file path for the sorted gtf by pressing the “Sorted GTF:” button. Now, sort the GTF by pressing the “Sort” button. While PrimerSeq is sorting your GTF the “Sort” button should now say “Sorting …”. When sorting is finished you should see the button text return to “Sort”.
Adding Gene IDs
Warning!
If you download a GTF fromUCSC, you will need to add
correct Gene IDs
If your GTF is also from UCSC you can then use Edit -> Add Genes to add correct gene IDs. A dialog will appear and require your original GTF and a kgXref file. You can obtain a kgXref file from UCSC by doing the following:
- Go to the UCSC table browser
- Select Genes and Gene Prediction tracks from the group dropdown
- Select UCSC Genes from the track dropdown
- Select kgXref from the table dropdown
- Make sure the output format is ‘all fields from selected table’
- Click ‘get output’
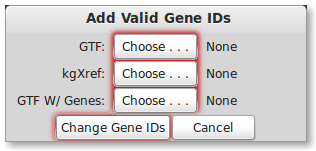
Please select your GTF file from UCSC by pressing the “GTF:” button. You will also need to select the kgXref file you downloaded from UCSC by pressing the “kgXref” button. Next, select the file name you wish to save your GTF as by pressing the “GTF W/ Genes” button. To start changing the gene IDs press the “Change Gene IDs” button. You will notice the button will be disabled while PrimerSeq is still in progress. When finished, the “Change Gene IDs” button will be available again.
Advanced Use at your own risk
PrimerSeq also allows you to mix GTF files together or use output from transcript assembly programs like Cufflinks. Like any normal GTF, output from programs like Cufflinks can be used as long as it is properly sorted (see above for details). In addition you can combine two or more GTF files into a single input GTF file for PrimerSeq. Besides sorting, the only additional action you must perform is to select “Not Valid” from the Gene ID dropdown in the Optional tab of the GUI. You need to specify the gene IDs as “Not Valid” because the two or more GTFs likely will not have the same gene ID even though they may have many of the same genes in their annotation.